
2019-05-30
Evolution of sequencing technology
Since the human genome was sequenced and famously published in Nature and Science in 2001(1)(2), sequencing technology has experienced significant advancement. In 2005 several high- throughput approaches, collectively referred to as Next Generation Sequencing (NGS), quickly became preferable for larger projects compared to the more traditional Sanger sequencing, a capillary electrophoresis-based method used widely for close to 40 years. NGS technology produces many very short overlapping reads simultaneously, using massively parallel sequencing technology, such that each section of DNA (or RNA) is sequenced multiple times for exceptionally high coverage. The introduction of NGS played a huge role in revolutionizing the genomics field by lowering the cost of genome sequencing and by providing results 100 times faster than the Sanger approach. In 2010, the emergence of Third-Generation Sequencing (TGS) - enabled sequencing from a single molecule of DNA, thus eliminating the need for amplification and reducing PCR-derived bias. TGS produces substantially longer reads at 10K-15K base pairs compared to 100-600 base pairs for NGS, resulting in increased quality of genome assemblies through higher consensus accuracy and more uniform sequence coverage. For perspective on the evolution of sequencing, the 13-year-long, $2.7-billion international project (Human Genome Project) that sequenced the first human genome back in 2001, today would take little more than a day and would cost around $1,500 using NGS.
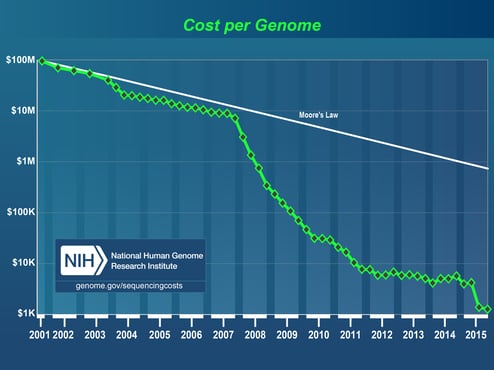
Figure 1: Cost of sequencing a human-sized genome. By the NHGRI Genome Sequencing Program.
Sequencing the microbiome
Until fairly recently, identification of bacterial species from a complex sample relied mainly on culture-based techniques. Because some bacteria are slow-growing or uncultivable, the limitations associated with bacterial culture pushed species detection towards the use of genetic markers. The most commonly used marker for the identification of bacteria is the 16S rRNA gene, which is present in all bacteria and consists of both highly conserved and hypervariable regions. The conserved regions are used for gene amplification (binding site for universal primers) whereas the interspersed hypervariable regions provide a way to discriminate between bacterial families, genera, and, in rare cases, species. Most studies have used targeted 16S rRNA NGS due to the low cost, fast turnaround, and the wide availability of sequence references in public and private databases. Despite its widespread use for microbiota profiling, the technique suffers from poor resolution below the bacterial genus level, making it difficult to differentiate closely related species. Due to the short reads generated by NGS technology, the sequence coverage is sparse and only partially covers the entire 1,550 base pair long 16S rRNA gene. Although the capability of TGS sequencing to provide reads covering the entire 16S rRNA gene in a high throughput manner mitigates some of the aforementioned limitations, the added cost over NGS sequencing may not be worth the benefit. There exists another alternative - shotgun metagenomics sequencing - that may provide an answer.
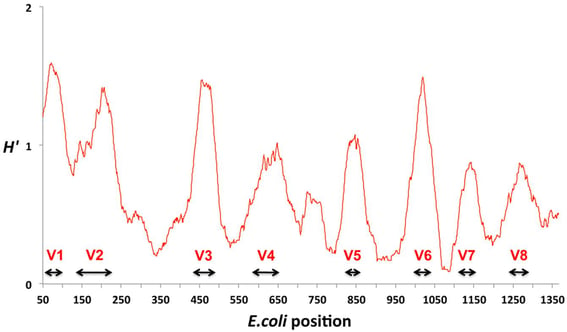
Figure 2: Hypervariable regions of the 16S rRNA gene (V1 to V8). Conservation is assessed using the Shannon entropy value (H′) where a non-variant nucleotide has an entropy of zero. Adapted from: Vasileiadis, Sotirios, et al. "Soil bacterial diversity screening using single 16S rRNA gene V regions coupled with multi-million read generating sequencing technologies." PloS one 7.8 (2012): e42671.
Shotgun Metagenomic Sequencing (SMS)
In contrast to 16S sequencing, which offers a phylogenetic survey on the diversity of a single gene, SMS has the ability to sequence the complete collection of microbial genomes present in a microbiome sample, theoretically being able to discriminate between strains that differ by a single nucleotide. In addition, because SMS does not rely on the amplification of a particular phylogenetic marker, organisms that were not known to be present or that lack a particular marker, such as fungi, phage and viruses, may be identified. SMS also provides greater detection sensitivity, as organisms are identified using their entire genomes rather than only the 16s rRNA gene. Although the benefits of using SMS over marker gene sequencing for microbial profiling seem obvious, the technology has some drawbacks. First, the cost to perform SMS is considerably greater than that of 16S sequencing, making SMS often not a plausible alternative for projects comprising a large number of samples. The second challenge facing SMS is the requirement for considerable bioinformatics and computational resources to make accurate interpretations from the large volume of raw data and this may not be feasible for all researchers. To add to this challenge, samples collected from sites where host DNA is present in significant amount (such as skin or oral microbiome samples) will require further bioinformatic filtering as well as higher sequencing depth. This is due to the SMS technology not being selective for only microbial borne signal, unlike gene targeted NGS. This post-sequencing bioinformatics analysis contributes to the elevated cost of SMS.
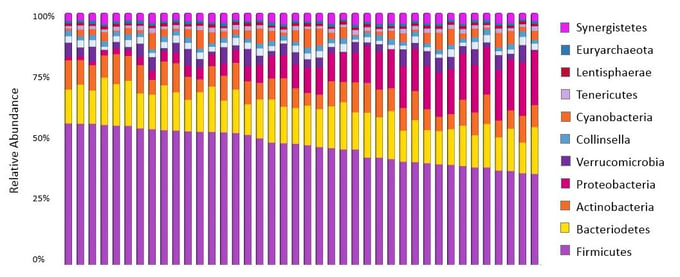
Figure 3: Percent abundance (relative) of the bacterial representation in the microbiota.
Figure from DNA Genotek
NGS or TGS? 16S or SMS?
Here we provide a brief overview of the evolution of sequencing technologies (NGS and TGS) and the predominant alternatives for microbiome profiling (16S rRNA and SMS). There are clearly more aspects to account for when considering a sequencing approach since many other factors impact the quality of the results, such as sequencing error rate, depth and coverage of sequencing, bioinformatics analysis pipelines, and reference database fidelity and completeness. Each microbiome study is different and investigators are seeking experimental designs that will generate the most robust and accurate scientific findings for the lowest sequencing cost possible. We’d love to hear your input with respect to your sequencing technology of choice. Also, where do you think the technology is headed in the future?
If you have any questions related to microbiome study design or our SMS or 16S rRNA sequencing and bioinformatics services, please contact us to learn about our microbiome services.

References:
1. Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G., ... & Gocayne, J. D. (2001). The sequence of the human genome. science, 291(5507), 1304-1351.
2. Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., ... & Funke, R. (2001). Initial sequencing and analysis of the human genome.