2023-07-10
When developing a clinical or diagnostic test or assay for patient use, a strong indicator of device effectiveness and safety is a recognized designation from the United States regulatory body, the Food and Drug Administration (FDA). Acknowledgement by the FDA is beneficial if the device in question is to be marketed to the U.S. population for human health purposes.
DNA Genotek, Inc. —a subsidiary company of OraSure Technologies, Inc.—is pleased to say that two of our saliva collection product families have now received FDA general use 510(k) clearance; these are the Oragene™•Dx and ORAcollect™•Dx devices. This designation demonstrates that our devices are recognized by the FDA for the collection and stabilization of human DNA, from saliva, to be used in prescription and/or over-the-counter (i.e., self-collected, at-home sampling) genetic testing applications.
FDA general use 510(k) clearance: when and how?
Back in January of 2020, DNA Genotek, a subsidiary company of OraSure Technologies, Inc., first received FDA general use 510(k) clearance for its Oragene™•Dx family of products. Oragene™•Dx devices are intended for the non-invasive collection of saliva samples for in vitro diagnostic testing of human DNA. The Oragene™•Dx saliva collection device was included as a component in the De Novo authorization granted by the U.S. FDA to Helix for their Helix® Laboratory Platform—a qualitative in vitro diagnostic whole exome sequencing platform that detects single nucleotide variants (SNVs) and small insertions and deletions (indels) in human genomic DNA. This was the first exome sequencing-based platform authorized by the FDA. The Oragene™•Dx device was also included as a component in the 510(k) clearance that Helix received for its Helix® Genetic Health Risk App for late-onset Alzheimer’s Disease for over-the-counter use—the first test to be cleared on the Helix® Laboratory Platform.
More recently, in November 2022, OraSure Technologies acquired the same designation for its ORAcollect™•Dx product family. OraSure Technologies received this designation through its partnership with Grifols who offer a test to screen for alpha1-antitrypsin deficiency (alpha-1) in individuals who may be at risk for alpha-1. Alpha-1 raises patients’ risks for lung disorders, and it is estimated that about 3% of COPD patients have alpha-1.
Both FDA-cleared saliva collection devices allow saliva samples to be stabilized for use in downstream diagnostic testing applications and can be transported and/or stored long-term at ambient temperatures. In separate usability studies, both devices have been demonstrated to simplify the sample collection process for naïve donors, providing ease-of-use and convenience with the self-collection aspect. User comprehension of the collection instructions with Oragene™•Dx and ORAcollect™•Dx devices was found to be over 95%, implying users found the device instructions easy to understand.
Save yourself time and money—use FDA-cleared devices
By leveraging pre-existing FDA-cleared saliva collection devices, diagnostic companies can save time and money. FDA general use 510(k) clearance allows genetic test providers/manufacturers to confidently and quickly incorporate the FDA-cleared saliva collection devices within their test protocols, in either a prescription or DTC model, where collection can be done by either a healthcare professional or the patient themselves. As these DNA Genotek devices have already received FDA clearance, providers can reduce the time and burden of resource investments by genetic testing/diagnostic companies in acquiring FDA clearance for the collection aspect of their tests.
The general clearance also allows manufacturing companies more flexibility with branding options including uniquely designed collection device packaging (secondary outer box), co-branding of device labels and user instructions, custom barcoding, and other collateral such as welcome letters and consent forms. 
For test manufacturers that are contemplating clearance now or may require FDA clearance dependent on outcomes of the Verifying Accurate Leading-edge in vitro clinical tests (IVCT) Development (VALID) Act, using FDA-cleared collection devices will save both time and money in the long run, rather than attempting to validate collection devices that are not FDA-cleared for general use.
First introduced in 2021, the VALID Act is an U.S. legislative bill that would establish a new category of in vitro clinical tests that includes test kits and laboratory-developed tests. If enacted as law, this bill would require that the FDA regulate (review and approve) in vitro clinical tests, pre-market. 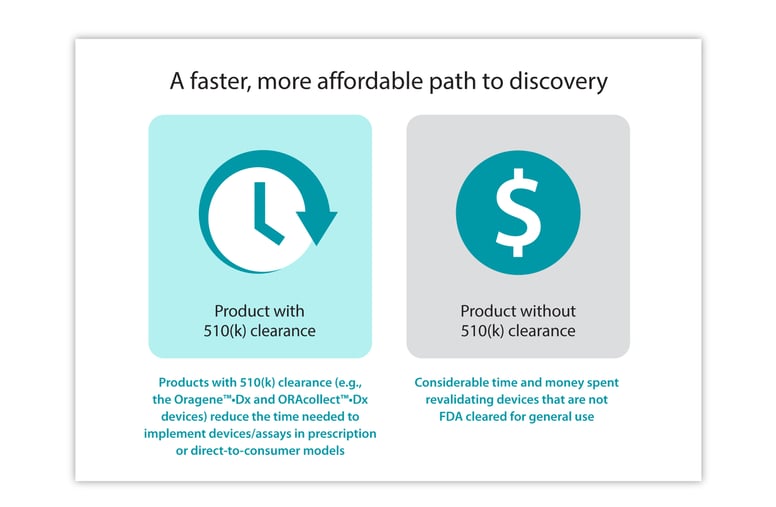
Trusted expertise, backed by a decade of experience and customer support
Apart from saving time and money, the FDA-cleared devices provided by DNA Genotek come with added perks! DNA Genotek offers several decades worth of experience and knowledge from supporting genetic test providers with their FDA clearances. Customer success is our priority, which is why we provide our customers with regulatory and product and marketing support for their own 510(k) submissions, offering a faster and more affordable path to clearance. We also offer professional customization, branding, and order fulfillment services to those interested in streamlining their sample workflow.
Quality you know you can count on—6,000 customers in over 100 countries
As an industry leader in product innovation, sample quality, and customer service, it is not surprising to see why ten of the top 10 U.S. hospitals, twenty of the top 20 global universities, and hundreds of genetic testing companies choose our products. To date, more than 6,000 customers in over 100 countries trust and use our products to enable their genetic testing or research projects.
If you would like to learn more about our Oragene™•Dx or ORAcollect™•Dx family of products or would like to evaluate these devices for yourself, please contact us at info@dnagenotek.com.