
2018-07-30
A critical component to any human gut microbiome study is the ability to accurately represent the ‘true’ microbial community present at a specific period in time or, in other words, a snapshot. With such a diverse sample type, there is considerable inter- and intra-subject variability, as well as methodological and technical sources of variation that can increase noise and obscure results. In this blog, we focus on discussing a technical source of variation – nucleic acid extraction methods.
Using culture-independent methods, the first step following fecal sample collection and stabilization in assessing microbial diversity is nucleic acid extraction. There are currently many commercial and homebrew extraction methods available, with differences in lysing procedure (i.e. mechanical bead-beating, chemical, heat or enzymatic methods), isolation methods (i.e. magnetic beads or column-based options), as well as differing platforms (i.e. manual versus automated). The aim, of course, is to have consensus on a universal standardized methodology across all gut microbiome research permitting meta-analysis and cross-study comparisons, as well as limiting a source of preventative bias. To achieve this auspicious goal, studies specifically evaluating the most accurate, reproducible and efficient means of microbial nucleic acid extraction methods are needed – thus allowing researchers to make informed decisions regarding their study design.
“The accuracy of microbiome data depends on how well the DNA is extracted from the gut bacteria, so that it accurately reflects the composition of the actual gut microbial community.”[1]
A good illustration of the variation between DNA extraction methods is shown in the recent publication by Mi Young Lim, Young-Do Nam and colleagues from the Korea Food Research Institute titled ‘Comparison of DNA Extraction Methods for Human Gut Microbial Community Profiling’. [1] In this study, the researchers used a fecal sample from a single donor collected into four OMNIgene•GUT kits that stabilized the samples at ambient temperature. The samples were then pooled and the pooled sample was used to evaluate three different commercial DNA extraction kits. The choice to only collect from a single donor and to pool the stabilized samples eliminates inter-individual variation and greatly reduces intra-individual variation. The effects of the DNA extraction methods were evaluated based on the quality and quantity of nucleic acids, as well as the gut microbial community composition assessed via 16S rRNA gene sequencing.
The three commercial extraction kits evaluated were:
- TianLong Stool DNA/RNA Extraction Kit (TS; Xi’an TianLong Science and Technology Co., Ltd., China);
- QIAamp DNA Stool Mini Kit (QS; Qiagen, Germany); and
- QIAamp PowerFecal DNA Isolation Kit (QP; Qiagen, Germany)
With these three kits, nine different extraction protocol variations were tested (See Figure 1). Unlike the QP kit, the TS and QS do not normally include a mechanical lysis step, so both the TS and QS kits were assessed with and without the addition of mechanical lysis by bead-beating. Furthermore, the TS and QS kits were compared using automated instruments, as well as manual methods. The QP kit was compared using only the manual method.
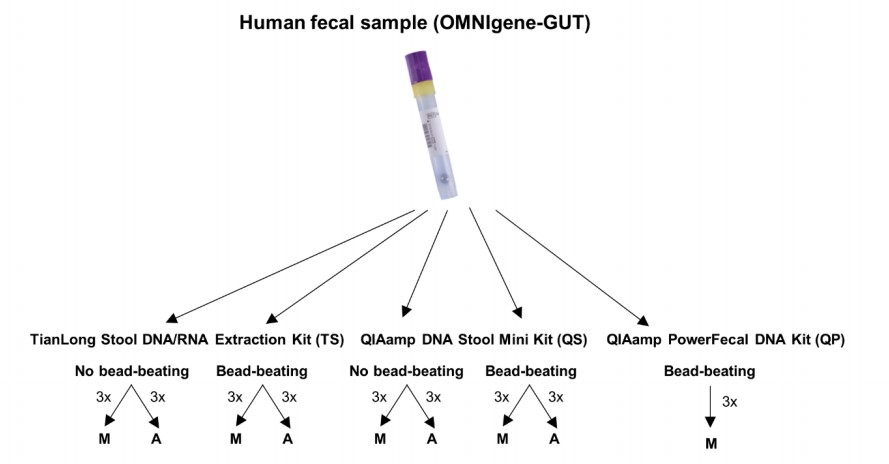
Figure 1. Schematic representation of the study design (Adapted from Supp Fig. 1 from1)
In assessing microbial DNA quantity: Although nucleic acid yields from all three extraction kits were sufficient to successfully accomplish 16S rRNA gene sequencing, the TS kit had significantly higher concentrations than both the QP and QS kits. This could have been due to its use of magnetic beads, while the other two kits use a silica filtration column.
In terms of DNA integrity: Regardless of the inclusion of bead-beating or the use of automated methods, QS and QP kits produced high molecular-weight DNA bands without smears visualized via agarose gel electrophoresis; meaning less DNA fragmentation. This finding means extracting DNA using QP and QS methods would be far better for single-molecule or long-read sequencing technologies than TS kits. TS kits might be appropriate for short-read sequencing technologies, but this limited utility for metagenomic library construction or shotgun metagenomics sequencing is a considerable drawback.
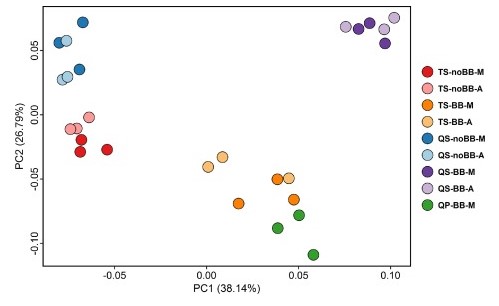
Figure 2. Principal coordinates analysis based on Bray-Curtis distances (Adapted from Fig. 3 of1)
Most importantly, in terms of species diversity, the methods that used bead-beating provided greater beta diversity than the non-bead-beating methods, as can be visualized in the clustering pattern present in the Bray-Curtis plot (Figure 2). Note that the sample replicates and automated/manual methods from the same extraction protocols are clustered in close proximity indicating their similarity. In the bead-beating methods, a greater abundance of gram-positive bacteria were present. This means that without adding mechanical disruption of the bacterial walls, the abundance of gram-negative bacteria may be over-exaggerated and not indicative of an accurate representation of the microbial community present. This finding highlights the fact that different extraction methods, those with and without bead-beating steps, can produce different results from the same sample.
In this study, we believe that the QP methodology with its inherent mechanical disruption step is the best extraction method. The nucleic acid yield was sufficient for downstream analysis, the DNA was high-molecular weight (which the TS method was not) and no additional bead-beating step needed to be added to the manufacturer’s protocol. There is a benefit to using an all-in-one commercial solution.
Furthermore, this study highlights the importance of carefully choosing an appropriate extraction method in microbiome studies. Different commercial kits, in the absence of inter- and intra-individual variation, exhibited different microbial community profiles due to technical variation.
If you would like to try the OMNIgene•GUT used in this article for your own evaluation, you can request samples below.
